




J. Walder, P. Kupková, V. Kaučák, K. Novobílský
Souhrn
Mitochondriopatie představují heterogenní skupinu systémových onemocnění, která vznikají na podkladě mutace nukleární či mitochondriální DNA. Prevalence dědičných mitochondriopatií se odhaduje na jeden případ na 5 000 narozených. Defekt mitochondriálního energetického metabolismu se typicky manifestuje výrazněji v orgánech s vyššími metabolickými nároky – v mozku, myokardu, játrech, v kosterním svalstvu či endokrinních orgánech, symptomatologie tak bývá velmi pestrá a srdeční postižení není specifické. Typická kardiální manifestace zahrnuje hypertrofickou a dilatační kardiomyopatii, arytmie – jak bradyarytmie, tak tachyarytmie, a srdeční selhání. Diagnostika mitochondriopatií je snadná pouze v případě typického neurologického a kardiologického obrazu, ale ve většině případů je obraz atypický a diagnóza obtížně stanovitelná. To dokumentuje i následující kazuistika, ve které popisujeme případ mladého muže, který byl primárně hospitalizován v Městské nemocnici Ostrava pro dekompenzovanou hypertenzi a až následující vyšetření a podrobná anamnéza postupně vedla k odhalení této vzácné diagnózy.
© 2019, ČKS.
Klíčová slova:
Hypertrofická a dilatační kardiomyopatie
Dekompenzovaná hypertenze
Mitochondriopatie
Abstract
Mitochondrial disease is a heterogeneous group of systemic diseases that develop consequent to mutations in nuclear or mitochondrial DNA. The prevalence of inherited mitochondrial disease has been estimated to be greater than 1 in 5,000 births. Defect of mitochondrial energetic metabolism is typically manifested in a more pronounced manner in organs with higher metabolic requirements – in the brain, myocardium, liver, muscles and endocrine organs, therefore symptomatology is so colourful and cardiac disability is not specific.
The typical cardiac manifestation of mitochondrial disease contains hypertrophic and dilated cardiomyopathy, arrhythmia – both bradyarrhythmia and tachyarrhythmia, and heart failure.
The diagnosis of mitochondriopathies is simple in case of typical neurological and cardiac picture, but in most cases the clinical features are atypical and diagnosis is difficult to determine, as documented in the following case report. This case report describes the case of a young man, who was primarily hospitalized in Municipal Hospital Ostrava with decompensated hypertension and gradually further examinations and look into his detailed medical history led to the revelation of this rare diagnosis.
Keywords:
Decompensated hypertension
Hypertrophic and dilated cardiomyopathy
Mitochondriopathies
Mitochondriální onemocnění patří mezi vzácné nemoci, což je dle European Union Regulation on Orphan Medicinal Products (1999) definováno jako onemocnění postihující jednoho člověka na 2 000 narozených v evropské populaci.1 Jedná se o heterogenní skupinu systémových onemocnění, která vznikají na podkladě mutace nukleární či mitochondriální DNA a mohou se manifestovat v kterémkoliv věku. Obecně se dá říci, že mitochondriální onemocnění vycházející z mutace mtDNA se častěji manifestují v dospělém věku, zatímco onemocnění způsobné mutací nDNA je častější u novorozenců a dětí.2
Prevalence dědičných mitochondriopatií se odhaduje na jeden případ na 5 000 narozených,3 dle dat Ústavu dědičných a metabolických poruch se minimální prevalence odhaduje na 1 : 3 400. Od roku 1992 bylo toto onemocnění v České republice diagnostikováno u přibližně 460 pacientů.4
První porucha mitochondriální funkce byla popsána Luftem v roce 1959.2 Od té doby bylo popsáno v mtDNA více než 200 bodových mutací, delecí a duplikací, které vedou k poruchám energetického metabolismu a rozvoji klinických příznaků onemocnění.4 Vzhledem k tomu, že mitochondrie jsou jediným zdrojem energie ve formě makroergních fosfátů pro buňku, manifestuje se onemocnění výrazněji v orgánech s vyššími metabolickými nároky – v mozku, myokardu, játrech, v kosterním svalstvu či endokrinních orgánech, symptomatologie tak bývá velmi pestrá.5
Některé poruchy postihují jen jeden orgán (např. oko u Leberovy hereditární optické neuropatie), obvykle však bývá postiženo několik orgánových systémů, nejčastěji nervový a muskuloskeletální, a proto se s touto skupinou onemocnění setkávají primárně ve svých praxích pediatři a u dospělých pacientů neurologové.6 Mezi nejčastější extrakardiální manifestace mitochondriopatií patří progresivní svalová slabost, retinitis pigmentosa, progresivní oftalmoplegie a encefalomyopatie.7
Kardiální postižení bývá časté, není však specifické. Zahrnuje kardiomyopatie, nejčastěji hypertrofickou a pak dilatační kardiomyopatii, arytmie – jak bradyarytmie, tak tachyarytmie, srdeční selhání, dilataci aortálního kořene, koronární postižení nebo náhlou srdeční smrt.8
Mitochondriální kardiomyopatie může být popsána jako postižení srdeční tkáně charakterizované abnormální strukturou srdečního svalu, funkcí nebo obojím, vzniklé v důsledku genetického defektu postihující mitochondriální respirační řetězce, při absenci konkomitantní ischemické choroby srdeční, hypertenze, chlopenní vady nebo vrozené srdeční vady.9
Na EKG bývá krátký interval PR, atrioventrikulární (AV) blokády, raménkové blokády často v kombinaci s obrazem hypertrofie levé komory. Zhanq a spol. retrospektivně analyzovali data (EKG, holterovský monitoring, echokardiografie a laboratorní výsledky) u 90 pacientů s diagnostikovanou mitochondriopatií. Z tohoto počtu pět pacientů mělo kardiomyopatii (dva hypertrofickou kardiomyopatií, tři pacienti dilatační kardiomyopatii) – prevalence byla 5,6 %.10 Dle práce českých autorů je prevalence kardiomyopatie u dětí ještě vyšší – v literatuře se uvádí až 40 %.11
Prevalence různých arytmií dosahovala 22,2 %. Dominující byly tachyarytmie – 14 pacientů, třem pacientům byl implantován trvalý kardiostimulátor pro bradyarytmii (diagnóza mitochondriopatie byla stanovena přibližně tři roky po implantaci).10
Na základě těchto údajů je patrné, že incidence kardiovaskulárních onemocnění u této skupiny onemocnění je vysoká a je prokázáno, že také mortalita je vyšší u pacientů se srdečním onemocněním (71 %) než bez něj (26 %), proto by každý mladý pacient s kardiomyopatií a závažnou převodní poruchou měl podstoupit genetické vyšetření a naopak pacienti se zjištěnou mitochondriopatií by měli podstoupit komplexní kardiologické vyšetření.12
V následující kazuistice popisujeme případ, kdy první manifestací tohoto onemocnění byla atypicky dekompenzovaná hypertenze.
Následující kazuistika popisuje případ 28letého muže, obézního habitu (BMI 40), s koktavostí od raného dětství, s lehkou mentální retardací, absolventa vyšší odborné školy, v nedávné době léčeného pro hypertenzi u svého praktického lékaře (délka léčby jeden rok), bez jiných dalších přidružených onemocnění.
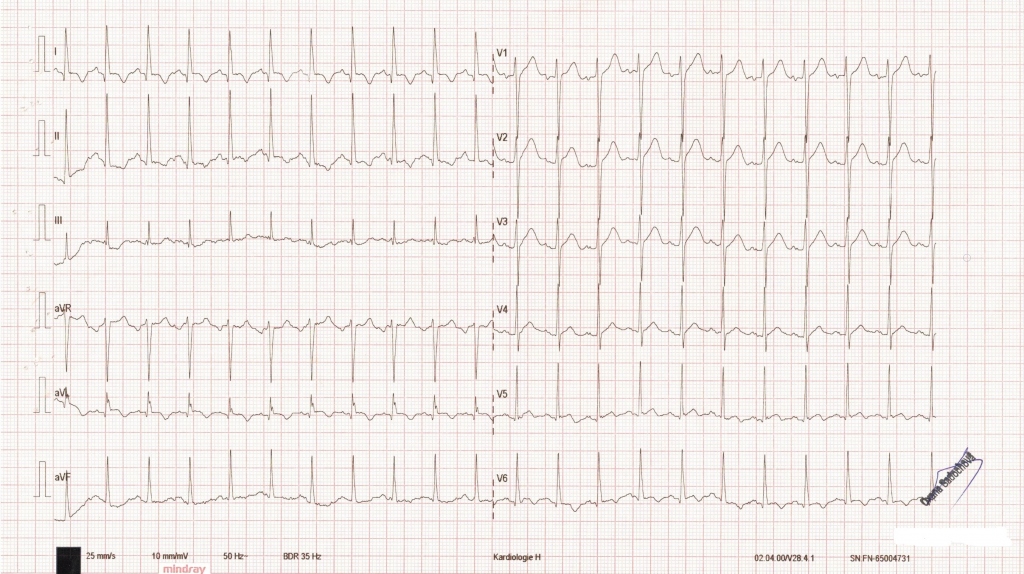
K hospitalizaci byl odeslán svým ošetřujícím lékařem pro dekompenzovanou hypertenzi s krevním tlakem (TK) 220/120 mm Hg, pacient udával jen slabost a únavu. Fyzikální vyšetření kromě obezity neprokázalo žádnou závažnou patologii. Při přijetí dominovala v laboratoři výrazná hypokalemie a hypomagnezemie. Na EKG byly patrné známky hypertrofie levé komory (LK), krátký interval PR, 1,5mm elevace úseku ST ve svodech I, aVL (obr. 1), pacient byl bez klinických příznaků akutního koronárního syndromu, vstupní hodnota vysoce senzitivního troponinu byla zvýšena (161 ng/ml), což jsme přisuzovali hypertenzi. V kontrolní laboratoři však došlo k významnému nárůstu hodnoty vysoce senzitivního troponinu (1 344 ng/ml), vzhledem k trvajícím EKG změnám (obr. 2) byl pacient akutně katetrizován s nálezem jen nevýznamných aterosklerotických změn na ramus interventricularis anterior.

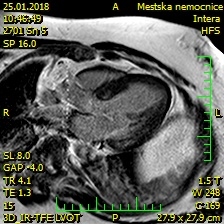
Dle zvyklosti byla doplněna echokardiografie, kde i přes omezenou vyšetřitelnost byla zjevná významná koncentrická hypertrofie stěn LK. K přesné verifikaci a ke zhodnocení etiologie hypertrofie byla provedena magnetická rezonance srdce, kde byla potvrzena významná koncentrická hypertrofie LK (max. 20 mm) s fokusy neischemické fibrózy v bazálním segmentu anterosepta a zadní stěny (obr. 3, 4). V rámci diferenciální diagnostiky hypertrofické kardiomyopatie a celkového habitu pacienta s mentální retardací jsme zvažovali dědičné metabolické choroby (např. Fabryho choroba), mitochondriální choroby, dědičné střádavé choroby (např. Danonova choroba) či komplexní syndromy (např. syndrom Noonanové, syndrom LEOPARD).

Následně bylo anamnesticky od rodičů zjištěno, že bratr pacienta zemřel ve věku 23 let na anesteziologicko-resuscitačním oddělení (ARO) Městské nemocnice Ostrava, kde byl došetřován pro suspektní mitochondriopatii (Kearnsův–Sayerův syndrom), nicméně před definitivním stanovením diagnózy zemřel na respirační selhání při septickém stavu, jeho další dvě sestry zemřely do dvou let věku bez udání příčiny.
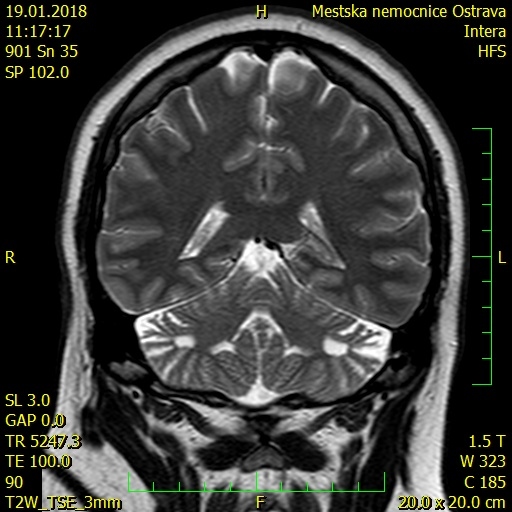
Na základě především rodinné anamnézy jsme tedy konzultovali Ústav dědičných a metabolických poruch. Dle doporučení bylo doplněno neurologické konzilium vstupně s normálním nálezem. Na magnetické rezonanci (MR) mozku (obr. 5) byl nález pseudocystických defektů obou mozečkových hemisfér (velmi suspektní v rámci mitochondriální poruchy), dále bylo provedeno oční vyšetření (bez patologického nálezu), endokrinologické vyšetření a MR srdce – viz výše. Výpočtem (HCM Risk-SCD calculator) bylo stanoveno riziko náhlé srdeční smrti 3,14, zatím tedy bez indikace implantace implantabilního kardioverteru-defibrilátoru (ICD).
Na pětikombinaci antihypertenziv byl pacient uspokojivě kompenzován. V průběhu hospitalizace jsme vyloučili nejčastější příčiny sekundární hypertenze (renální onemocnění – renovaskulární, renoparenchymatózní, primární hyperaldosteronismus, Cushingův syndrom, feochromocytom). Telemetricky nebyla zachycena žádná arytmie. Pacient byl propuštěn do ambulantní péče, kde bylo provedeno kontrolní endokrinologické vyšetření s nálezem centrálního hypogonadismu, dále byl objednán ke komplexnímu genetickému vyšetření do Všeobecné fakultní nemocnice v Praze. Dle předběžných výsledků byl průkaz delece mtDNA v moči.
V dalším průběhu onemocnění byl pacient v červnu 2018 hospitalizován na neurologickém oddělení Městské nemocnice Ostrava pro odeznělý amentní stav, kde již byla objektivizovaná zevní oftalmoplegie. Následně v říjnu 2018 byl přijat na ARO pro respirační selhání při bronchopneumonii, kde zmírá dle provedené pitvy na plicní embolii.
Tabulka 1 – Velká a malá diagnostická kritéria3
|
|
Velká diagnostická kritéria |
Malá diagnostická kritéria |
|
Klinická |
Mitochondriální syndrom nebo postižení jakýchkoli tří z následujících systémů: neurologický, muskulární, kardiální, renální, nutriční, jaterní, endokrinní, hematologický, ušní, oční, kožní nebo malformace, anamnéza mutací mtDNA nebo vyloučení jiné alternativní diagnózy |
Symptomy kompatibilní s defekty respiračního řetězce |
|
Histologická |
> 2 % červeně zbarvených vláken v kosterních svalech |
1–2 % červeně zbarvených vláken u pacientů mezi 30 a 50 lety, jakákoli červeně zbarvená vlákna, červeně zbarvená vlákna u pacientů mladších 30 let, více než 2% akumulace mitochondrií subsarkolemálně pod plazmatickou membránou u pacientů mladších 16 let, další elektronové abnormality v jakékoli tkáni |
|
Enzymatická |
> 2 COX-negativní vlákna u pacientů mladších 50 let, > 5 % COX-negativních vláken u pacientů ≤ 50 let, < 20 % činnosti jakéhokoli komplexu RC ve tkáni, < 30 % činnosti jakéhokoli komplexu RC v buněčné linii nebo < 30 % činnosti stejného komplexu RC aktivního ve dvou a více tkáních |
Manifestace vady v expresi komplexu respiračního řetězce založené na protilátkách, 20–30 % činnosti jakéhokoli komplexu RC v tkáni, 30–40 % činnosti jakéhokoli komplexu RC v buněčné linii nebo 30–40 % činnosti stejného komplexu RC aktivního ve dvou a více tkáních |
|
Funkční |
Snížená syntéza ATP fibroblasty o více než tři směrodatné odchylky pod průměrem |
Snížená syntéza ATP fibroblasty 2–3 směrodatné odchylky pod průměrem nebo fibroblasty nejsou schopny růst v prostředí s glukózou nahrazenou galaktózou |
|
Molekulární |
Identifikace mutace nDNA nebo mtDNA nesporné patogenicity |
Identifikace mutace nDNA nebo mtDNA nesporné, u mtDNA pravděpodobné patogenicity |
|
Metabolická |
|
Jedna nebo více známek snížené funkce RC |
ATP – adenosintrifosfát; COX – cytochromoxidáza; mtDNA – mitochondriální DNA; nDNA – jaderná DNA; RC – respirační řetězec.
Primární mitochondriopatie jsou klinicky, biochemicky a geneticky velice heterogenní skupina, proto jejich diagnostika bývá často obtížná. Velice důležitou úlohu v těchto případech hraje rodinná anamnéza s výskytem systémového onemocnění, nicméně ne všechny osoby s mutací mtDNA se musí systémovým onemocněním manifestovat.
Hlavní diagnostická kritéria se opírají o kombinaci klinických, histologických, enzymatických, funkčních a molekulárních faktorů (viz tabulku 1),3 přičemž dvě velká kritéria nebo jedno velké a dvě malá kritéria stanovují diagnózu jako definitivní, jedno velké a jedno malé kritérium nebo tři malá kritéria jako pravděpodobnou, jedno velké nebo dvě malá kritéria jako možnou.
Klinická manifestace zahrnuje obvykle současné postižení několika orgánových systémů (viz tabulku 2).3 Nejčastěji bývá postižen nervový systém (přibližně 45 % pacientů), přibližně 20 % pacientů má intelektuální postižení nebo psychiatrické onemocnění, časté je postižení endokrinního systému (diabetes mellitus, diabetes insipidus, hypogonadimus,…) a kardiologické, méně časté pak renální či hematologické onemocnění.2
V případě podezření na mitochondriopatii je zlatým standardem ve stanovení diagnózy svalová biopsie, nicméně specifita ani senzitivita není 100%, histologický nález může být v některých případech dokonce normální. Endomyokardiální biopsie není rutinně doporučována, protože nejsou stanoveny hraniční hodnoty pro srdeční sval. Dle práce autorů Meyers a spol. pouze 13 % pacientů se známou mitochondriální poruchou mělo ultrastrukturální abnormality.3
Z hlavních diagnostických kritérií se pak v našich podmínkách dále provádí molekulárněgenetické vyšetření s průkazem mutace nDNA či mtDNA nesporné patogenicity.
V našem případě jsme se tedy v rámci diagnostiky opírali o několik faktorů – hlavní úlohu hrála rodinná anamnéza s vysoce pravděpodobným mitochondriálním onemocněním (Kearnsův–Sayerův syndrom u bratra), postižení kardiálního systému (hypertrofie LK) společně s mentální retardací, hypogonadismem při normálním nálezu na hypofýze a následně pak progresivní oftalmoplegie a dále průkaz mutace mtDNA v moči. Pacient tedy splňoval dvě hlavní diagnostická kritéria. Svalová biopsie nebyla provedena.
Prognózamitochondriálních onemocnění s výjimkou některých poruch beta-oxidace mastných kyselin není obvykle příznivá. Průběh onemocnění je často progresivní a léčba pouze symptomatická. Přestože u většiny postižených není významně ovlivněn klinický průběh onemocnění, stále se většina odborníků shoduje v názoru, že by se u postižených jedinců mělo vyzkoušet podávání L-karnitinu, koenzymu Q10 nebo cytochromu C, v některých situacích se doporučuje podávat rovněž L-arginin, riboflavin, vitamin K3, thiamin, vitamin C a E.4
Důležitá je především mezioborová spolupráce a důsledná práce s pacientem a jeho rodinou.
Tabulka 2 – Klinická manifestace postižení různých orgánových systémů3
|
Systém |
Klinické projevy |
Systém |
Klinické projevy |
|
Kardiovaskulární |
Srdeční selhání |
Muskuloskeletální |
Svalová slabost s normální hodnotou kreatinkinázy a normální EMG |
|
Arytmie |
Malý vzrůst |
||
|
Šelest |
Mikrocefalie |
||
|
Náhlá srdeční smrt |
Kulatý obličej |
||
|
Non-kompaktní kardiomyopatie |
Vysoké čelo |
||
|
Apikální balonkový syndrom |
Nízce postavené uši |
||
|
Respirační |
Dušnost |
Krátký krk |
|
|
Ortopnoe |
Kůže a měkké tkáně |
Hypertrichóza |
|
|
Respirační selhání |
Ekzém |
||
|
Respirační acidóza |
Vitiligo |
||
|
Neurologický |
Encefalopatie |
Lipomatóza |
|
|
Ataxie |
Retikulární pigmentace |
||
|
Pohybové postižení |
Gastrointestinální |
Parodontóza |
|
|
Záchvaty |
Anorexie |
||
|
Mentální retardace |
Bolesti břicha |
||
|
Renální |
Selhání ledvin |
Nauzea |
|
|
Benigní renální cysty |
Zvracení |
||
|
Fokální segmentální glomeruloskleróza |
Průjem |
||
|
Proximální tubulopatie |
Malabsorpce |
||
|
Nefritický syndrom |
Atrofie klků |
||
|
Tubulointersticiální nefritida |
Zácpa |
||
|
Hematologický |
Diabetes mellitus |
Pseudo-obstrukce |
|
|
Diabetes insipidus |
Pankreatitida |
||
|
Hypofunkce štítné žlázy |
Zvýšená aktivita jaterních enzymů |
||
|
Hypoparathyreoidismus |
Oční |
Externí oftalmoplegie |
|
|
Deficit ACTH |
Retinitis pigmentosa |
||
|
Hypogonadismus |
Sluchový |
Senzorineurální ztráta sluchu |
|
|
Amenorea |
|||
|
Gynekomastie |
ACTH – adrenokortikotropní hormon; EMG – elektromyografie.
Mitochondriopatie představují skupinu vzácných onemocnění, jejichž diagnostika bývá velmi obtížná pro jejich pestrou symptomatologii. Přestože kardiální postižení bývá časté, není pro tuto diagnózu specifické. Typická kardiální manifestace zahrnuje hypertrofickou a dilatační kardiomyopatii, arytmie a srdeční selhání, proto by měl každý mladý pacient s nově zjištěnou kardiomyopatií a závažnou převodní poruchou podstoupit genetické vyšetření a naopak pacienti se zjištěnou mitochondriopatií by měli podstoupit komplexní kardiologické vyšetření. Na základě výše uvedené kazuistiky bychom pak chtěli zdůraznit, jak je důležité provést komplexní vyšetření u mladých pacientů s nově zjištěnou hypertenzí, především důsledný odběr anamnestických dat se zaměřením na rodinnou historii, obzvláště při současném nálezu kardiomyopatie a lehké mentální retardace, protože i za touto běžnou diagnózou se může vyskytovat vzácné onemocnění.
Prohlášení autorů o možném střetu zájmů
Autoři prohlašují, že nemají žádný střet zájmů.
Prohlášení autorů o etických aspektech publikace
Rukopis dosud nebyl publikován v tištěné nebo elektronické podobě a není posuzován k publikování v jiném tištěném nebo elektronickém médiu. Všichni autoři přečetli a schválili konečnou verzi článku.
Popisovaná práce byla provedena v souladu s Etickým kodexem Světové lékařské asociace (World Medical Association) (Helsinskou deklarací).
Informovaný souhlas
Autoři prohlašují, že se požadavky na informovaný souhlas nevztahují na tento rukopis.
Literatura
V roce 2012 MUDr. Jakub Walder absolvoval obor všeobecné lékařství na Lékařské fakultě Univerzity Palackého v Olomouci. Od promoce pracuje na Kardiologickém oddělení Městské nemocnice v Ostravě. Od roku 2015 je členem České kardiologické společnosti. V současné době se připravuje k atestaci z oboru kardiologie.


